











请先登录您的atyun账户,方可使用该功能

审核通过后即可使用此功能,请耐心等待~






机器学习预测分子结构和电子特性,准确性高且成本低
2018年08月10日 由 浅浅 发表
143313
0
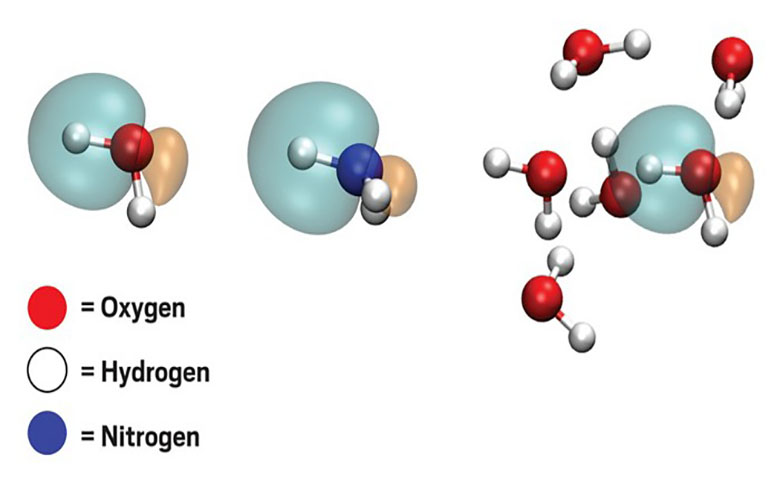
化学家想要模拟原子或分子的结构和电子特性时,他们常常转向叫做密度泛函理论(DFT)的计算技术。对于大多数用途,DFT可以在不需要太多计算能力的情况下产生准确的结果。当DFT失败时,化学家使用耦合聚类(CC)或Møller-Plesset扰动(MP2)理论等方法。这些产生比DFT更可靠的值,但是需要数倍于DFT的计算能力,即使对于小分子也是如此。
加利福尼亚理工学院的Thomas F. Miller及其同事现在证明机器学习可能是最好的方法,与CC或MP2一样准确,并且比DFT成本低。
研究人员希望预测电子结构相关能量,电子之间相互作用的量度,帮助化学家模拟分子的行为方式。机器学习方法基于一组已知数据预测这些值。
当计算化学家以前尝试用机器学习算法取代传统的计算技术时,他们已经根据分子中的原子类型或键角来训练系统。小组仅在一组小分子的局域分子轨道上训练他们的算法。
由于分子轨道对于潜在的键和原子是不可知的,Miller称新算法可以用一小组起始数据预测许多不同分子的性质。“随着系统规模的增长,这些大量的机器学习变量不再随着系统规模的增长而增加,我们可以用机器学习这些分子轨道,它们在不同的系统中是非常相似的。这使得机器学习变得不那么复杂。”
在一个例子中,研究人员在水的分子轨道上训练他们的算法,然后预测氨,甲烷和氟化氢的相关能量。对于甲烷,算法的值仅比CC生成的值低0.24%,这是他们发现的三个最不准确的结果。该团队的算法还比使用CC更快地预测相关能量。例如,对于一组六个水分子的计算需要2分钟的机器学习,而CC为28小时。
但该团队确实找到了算法不足的例子。在对甲烷和乙烷进行培训后,他们的系统在预测丁烷和异丁烷的值方面表现不佳。在训练集中包括丙烷可以获得更准确的结果。
Miller表示,相同的技术可用于预测各种分子的其他性质。他希望这种技术能够补充其他机器学习和电子结构技术,而不是替代它们。他强调说,这些早期结果距离任何人都可以使用的系统还有很长的路要走
加州大学欧文分校的计算化学家Kieron Burke认为,“这是一个很好的想法,看起来很有希望,但它可能比想象中更难以成为一种通用工具。”
论文:pubs.acs.org/doi/10.1021/acs.jctc.8b00636

欢迎关注ATYUN官方公众号
商务合作及内容投稿请联系邮箱:bd@atyun.com








广告

写评论取消
回复取消